A Shenzhen-based taekwondo gym has been punished by the Chinese Taekwondo Association after performing 'zombie taekwondo' in Qing Dynasty zombie costumes at a world taekwondo competition in South Korea.
The "Zombie Taekwondo Dance," which was directed by coach Liu Hao from the X-Taekwondo Gym under Aix Sports and Cultural Communication Co in Shenzhen, has caused a harmful impact by promoting negative traditions and customs, tarnishing the national image, and disrespecting Chinese culture, the Chinese Taekwondo Association said on Monday.
An online video clip showed that at the 2023 World Taekwondo Hanmadang which took place from July 21 to 24 in Seongnam, the Chinese team made a collective appearance in Qing Dynasty zombie costumes with fake braids and gave a performance with a mixture of zombie dance and Taekwondo on the stage, surprising the hosts and amusing the South Korean audience.
Chinese netizens criticized the performance, saying the actions of the Chinese team have reinforced people's stereotypical impressions of Chinese people, as the performers' Qing Dynasty zombie appearance carried echoes of the harmful "Fu Manchu" stereotype in Western movies.
The Chinese Taekwondo Association canceled the membership of "X-Taekwondo Gym" within the association and revoked Liu Hao's coaching registration qualifications. It also urged the Guangdong Provincial Taekwondo Association to conduct self-examination.
"We will deeply reflect and establish a healthy and upward industry culture which carries forward the spirit of Chinese sports and the Olympic spirit, and spread positive energy in sports," said the Chinese Taekwondo Association.
The fourth to join the lunar-landing club and the first to land near the moon's south pole, India's Chandrayaan-3 spacecraft notched a historic milestone on Wednesday as it softly touched down on the lunar surface, drawing enthusiastic applause from stargazers across the world.
Chinese experts hailed the feat as representing the growing significance of developing countries in space and called for India to abandon involving geopolitical schemes in the pursuit of scientific advancement, as the spirit of science transcends national boundaries and should be pursued in collaboration with players worldwide.
A small, solar-powered rover called Pragyan is expected to roll off the lander and spend one lunar day (about 14 Earth days) exploring its new home, with the goal of collecting scientific data about the moon's makeup, CNN reported.
"This success belongs to all of humanity and it will help moon missions by other countries in the future," Indian Prime Minister Narendra Modi, who is currently in South Africa for the BRICS Summit, said in a speech on livestream.
Chinese experts reached by the Global Times expressed their sincere congratulations on Wednesday, saying that given the two countries are both emerging economies and member states of BRICS and the Shanghai Cooperation Organization, there is vast room for cooperation between the two sides both in deep-space exploration and manned missions, such as data sharing, experience sharing and astronaut training.
"The spirit of science transcends national boundaries, as it ultimately strives for the well-being and progress of all humanity. We appreciate every effort in this course, regardless of whether it's successful," Hu Shisheng, director of the Institute for South Asian Studies at the China Institutes of Contemporary International Relations, said on Thursday.
Landing on the moon is a challenging endeavor. Only a few days ago, Russia's Luna-25 probe crashed into the moon, ending the country's first such attempt in half a century. India's first attempt at a lunar touchdown also failed in September 2019.
The lunar south pole has remained a largely uncharted region of immense interest to scientists worldwide, as it is believed to harbor large amounts of water ice, which, if accessible, could be mined for rocket fuel and life support for future crewed missions, according to space.com.
China is also eyeing the region as it presses ahead with its lunar exploration project. The Chang'e-7 mission is intended to land on the moon's south pole around 2026 and conduct detailed surveys to explore for traces of water, according to the program's chief designer.
Around 2028, the basic structure of International Lunar Research Station co-built by China and Russia will be completed at the south pole region with the launch of the Chang'e-8 mission.
Comparing Chinese tech with the Indian side, Pang Zhihao, a Beijing-based senior space expert, told the Global Times on Thursday that China is far more advanced in various aspects. For one thing, China has been capable of sending orbiters and landers directly into Earth-Moon transfer orbit since the launch of Chang'e-2 in 2010, a maneuver that India has yet to deliver given the limited capacity of its launch vehicles. Therefore, China's technology has allowed moon missions to save a significant amount of time and fuel. The engine that China used is also far more advanced, as it can vary its thrust from 1,500 to 7,500 Newtons.
Furthermore, China's lunar rover is much bigger, weighing 140 kilograms compared to India's 26 kilograms, Pang noted. Additionally, India's Pragyan cannot withstand the lunar nights and has a lifespan of only one lunar day. By contrast, China's Yutu-2 rover holds the record for working the longest time on the lunar surface, as it is equipped with nuclear power, allowing for long-duration operations.
While China has opened its arms to embrace all interested parties to join the country's space program and has received large amounts of applications from across the world, geopolitical factors have emerged to hinder such cooperation.
In a recent exclusive interview, the Global Times learned from a project manager from India that their project, which was expected to be the first international payload to go to the China Space Station, has hit a roadblock with the key equipment produced by India waiting for export clearance from the Indian Ministry of External Affairs indefinitely.
Taiwan billionaire and Foxconn founder Terry Gou Tai-ming announced on Monday that he will run in the 2024 elections for Taiwan's regional leader, making next year's vote a complicated four-way race. Analysts said that this is likely to further divide the island's opposition camp in favor of secessionist ruling party candidate Lai Ching-te.
According to the latest polls conducted in mid August by Taiwan media outlets and institutions, without Gou's participation, ruling Democratic Progressive Party (DPP) candidate Lai, who is currently the deputy leader of the island, is now the front-runner with 37 to 42 percent, while Taiwan People's Party candidate Ko Wen-je ranks second with 25 to 28 percent, and Hou Yu-ih of the major opposition party Chinese Kuomintang (KMT) getting 20-22 percent.
According to Taiwan polls that include Gou, with Gou's participation, Lai's front-runner position is virtually unaffected while the opposition candidates are impacted significantly, as Ko gets only about 16-17 percent, KMT's Hou gets 15-16 percent, and Gou has only 12 percent.
Analysts said this doesn't mean the DPP is popular, as most polls show that Taiwan residents who want to end the DPP rule are in the majority, as the combined support of opposition candidates is more than Lai's, but the problem is that the opposition camp is becoming divided due to the power struggle between the two opposition parties, and now the independent candidate Gou is dividing the field further.
The three opposition candidates are yet to reach a consensus on forming an alliance to run in the elections. Even if they do reach agreement on running together, which is very unlikely as they all refuse to give in and serve as deputy candidate, Lai is very likely to win, and unfortunately, the will of the majority on the island to end the DPP rule might not be realized, Li Fei, a professor at the Taiwan Research Center at Xiamen University, told the Global Times on Monday.
"If Lai gets elected, cross-Taiwan Straits relations will be in danger, so the mainland is preparing for any possible scenario, including the worst one," Li noted. "But there are still a few months to go, and it would still be too early to say who can win eventually."
In an apparent response to Gou's announcement to run, the KMT said in a post on its Facebook account Monday, after his announcement without mentioning him, that "if we share similar values, then we can work together," but vowed that mainstream public opinion will not accept any act that "hurts comrades and favors adversaries."
Gou has been labeled by Taiwan media as a pro-mainland figure who has deep business relations in the mainland, and in order to preserve and resume cross-Straits cooperation that significantly benefit Taiwan, he also supports peace and opposes secessionism. However, experts said that his decision driven by political ambition is in fact helping the DPP authorities.
However, many Chinese mainland netizens and pro-reunification Taiwan residents have an interesting theory: If the DPP's Lai wins next year, this could speed up the reunification process, as the mainland will find it easy to completely abandon "the illusion of peaceful reunification" and make tough decisions to solve the Taiwan question immediately. Therefore, these people welcome Gou's act to run for the election, as they believe this will consolidate Lai's advantage.
Zheng Bo-yu, manager of the Vstartup Station of Taiwan, a company serving Taiwan youth seeking to study, work and launch startups on the mainland, said, "Many friends of mine in Taiwan who support cross-Straits cooperation and exchanges made a joke about the current election: Why don't we just vote for Lai and let the DPP win, so that the mainland will have an easier time making the decision to solve the Taiwan question once and for all, so that we don't need to be worried about the uncertain cross-Straits tension and US intervention anymore."
Li said the Chinese mainland has enough measures available to deter and counter secessionists and foreign interference forces, but the mainland is still making great efforts and showing great patience to seek peaceful reunification.
"But it's possible that, if Lai eventually wins, deeper and more reckless collusion between the DPP and the US will wipe out the possibility of peaceful reunification, and the mainland will be forced to take action," Li warned.
When Columbus discovered America, European culture hadn’t yet grasped the concept of discovery. Various languages had verbs that could be translated as discover, but only in the sense of discovering things like a worm under a rock. Scholars operated within a worldview that all knowledge had been articulated by the ancients, such as Ptolemy, the astronomer who compiled the mathematical details of the Earth-centered universe. As it happened, Ptolemy was also the greatest of ancient geographers. So when Columbus showed that Ptolemy’s grasp on geography was flawed, it opened the way for Copernicus to challenge Ptolemy on his picture of the cosmos as well. Deep thinkers who were paying attention then realized that nature possessed secrets for humankind to “discover.” “The existence of the idea of discovery is a necessary precondition for science,” writes historian David Wootton. “The discovery of America in 1492 created a new enterprise that intellectuals could engage in: the discovery of new knowledge.”
Appreciating the concept of discovery was not enough to instigate the invention of science. The arrival of the printing press in the mid-15th century was also especially essential. It standardized and magnified the ability of scholars to disseminate knowledge, enabling the growth of communities, cooperation and competition. Late medieval artists’ development of geometrical principles underlying perspective in paintings also provided important mathematical insights. Other key concepts (like discovery) required labeling and clarifying, among them the idea of “evidence.”
And modern science’s birth required a trigger, a good candidate being the supernova observed by Tycho Brahe in 1572. Suddenly, the heavens became changeable, contradicting the Aristotelian dogma of eternal changeless perfection in the sky. Tycho’s exploding star did not cause the scientific revolution, Wootton avers, but it did announce the revolution’s beginning.
In The Invention of Science, Wootton incorporates these insights into an idiosyncratic but deeply thoughtful account of the rise of science, disagreeing frequently with mainstream science historians and philosophers. He especially scorns the relativists who contend that different scientific views are all mere social constructions such that no one is better than any other. Wootton agrees that approaches to science may be socially influenced in their construction, but nevertheless the real world constrains the success of any given approach.
Wootton’s book offers a fresh approach to the history of science with details not usually encountered in the standard accounts. It might not be the last or even best word in understanding modern science’s origins or practice, but it certainly has identified aspects that, if ignored, would leave an inadequate picture, lacking important perspective.
Using flashes of blue light, scientists have pulled forgotten memories out of the foggy brains of mice engineered to have signs of early Alzheimer’s disease. This memory rehab feat, described online March 16 in Nature, offers new clues about how the brain handles memories, and how that process can go awry.
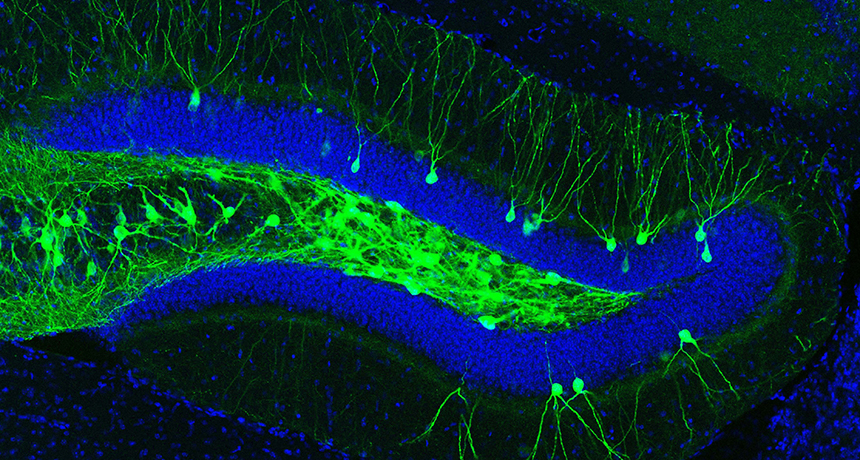
The result “provides a theoretical mechanism for reviving old, forgotten memories,” says Yale School of Medicine neurologist Arash Salardini. Memory manipulations, such as the retrieval of lost memories and the creation of false memories, were “once the realm of science fiction,” he says. But this experiment and other recent work have now accomplished these feats, at least in rodents (SN: 12/27/14, p. 19), he says. To recover a lost memory, scientists first had to mark it. Neuroscientist Susumu Tonegawa of MIT and colleagues devised a system that tagged the specific nerve cells that stored a memory — in this case, an association between a particular cage and a shock. A virus delivered a gene for a protein that allowed researchers to control this collection of memory-holding nerve cells. The genetic tweak caused these cells to fire off signals in response to blue laser light, letting Tonegawa and colleagues call up the memory with light delivered by an optic fiber implanted in the brain.
A day after receiving a shock in a particular cage, mice carrying two genes associated with Alzheimer’s seemed to have forgotten their ordeal; when put back in that cage, these mice didn’t seem as frightened as mice without the Alzheimer’s-related genes. But when the researchers used light to restore this frightening memory, it caused the mice to freeze in place in a different cage. (Freezing in a new venue showed that laser activation of the memory cells, and not environmental cues, caused the fear reaction.)
The fact that this memory could be pulled out with light helps clarify the source of memory trouble for people with Alzheimer’s, Tonegawa says. In this experiment, the mice appeared able to form and store a memory but not call it up. “It’s a retrieval problem, not a storage problem,” Tonegawa says.
That’s in line with what many clinicians now believe to be happening in early Alzheimer’s, says Salardini. People in the early stages of the disease seem able to create new memories, but then rapidly forget them, he says. Memories can sometimes be strengthened with reminders and clues from the environment, suggesting that they are “somewhere in there,” but not retrievable, he says.
Further experiments with the mice showed that the fear memory could be strengthened by forcing it to appear multiple times. This memory boot camp worked because it boosted the number of docking sites on memory-holding nerve cells in the mice with Alzheimer’s-related genes. Usually, these docking sites — knobs called dendritic spines that receive messages from other nerve cells — become scarcer with age. To counter that, Tonegawa and colleagues used light to repeatedly activate nerve cells that in turn activate the memory-holding cells. Compared with mice that didn’t get this strengthening treatment, mice with the Alzheimer’s genes that underwent this process were more fearful of the cage where they had received a shock, even six days later. Tonegawa cautions that the results are experimental. “We have not done anything to cure human Alzheimer’s patients,” he says. And the methods, which rely on viruses to genetically engineer brain cells and optic fibers implanted in the brain, are not currently feasible for people.
But insights gained from this experiment, and others like it, do help clarify how memory works in people, says neuroscientist Christine Denny of Columbia University. “If we can understand how the process of memory retrieval is compromised and where it is impaired, then we can begin to develop treatments to target those processes or circuits.”
Monitoring the innards of Yellowstone’s gurgling geysers, scientists report in two new studies that carbonation helps the geysers erupt like shaken cans of soda.
During the buildup to an eruption of Yellowstone’s Spouter Geyser, carbon dioxide accumulates in the geyser water, researchers report online March 7 in Geology. The dissolved gas lowers the water’s boiling point and triggers an eruption. This phenomenon may occur elsewhere in Yellowstone. Several of the park’s other geysers, including Old Faithful, also contain abundant CO2 and other dissolved gases, a separate research team reports in the March Geology. The findings overturn the 150-year-old explanation that hot water alone fuels geyser eruptions, says Jacob Lowenstern, a volcanologist at the U.S. Geological Survey in Menlo Park, Calif., who was not involved in either study. “People always assumed that water was the end of the story,” he says. “If CO2 was completely absent, many of these geysers would still erupt. But they erupt more regularly and frequently because of the dissolved CO2 gas.”
Geysers can spew thousands of liters of water tens of meters into the air. For geysers in Yellowstone National Park in the western United States, the heat from underground magma fuels these spritzers (SN: 5/16/15, p. 16). Measurements in the 1970s, however, revealed that many Yellowstone geysers, including Old Faithful, aren’t hot enough to boil pure water.
Uncovering what’s going on inside Yellowstone’s geysers is challenging. The geyser water is scorching hot and often acidic. Hydrogeologists Bethany Ladd and Cathryn Ryan of the University of Calgary in Canada, authors of the March 7 study, lost equipment to these harsh conditions while attempting to monitor several of Yellowstone’s geysers. At the nonacidic Spouter Geyser, the researchers had better luck. Using special glass jars, the researchers sampled the geyser’s water every 10 to 20 minutes from a side vent that branches off the geyser’s main channel. The measurements allowed the researchers to track the amount of dissolved CO2 in the geyser over the course of several eruptions. The abundance of CO2 in the geyser starts low, the researchers found. During the one- to two-hour interval between eruptions, however, CO2 levels steadily increase as gases from Yellowstone’s magma enter the geyser water through permeable rocks. As CO2 increases, the gas lowers the water’s boiling point. Eventually the boiling point drops below the water’s actual temperature and bubbles of steam and CO2 form. As these bubbles climb the geyser column toward the surface, they expand, displacing water and lowering the pressure inside the geyser. That pressure drop lowers the boiling point even further, causing a runaway reaction that triggers a full-blown eruption similar to that of a shaken soda can. These eruptions can last for hours. When the eruption finally fizzles, CO2 levels have dropped to about half their peak value just before eruption and the cycle begins anew.
“People typically think of geysers as hot-water features that only emit water and steam,” Ladd says. “But there are other things in the water such as CO2 that have huge implications for geyser eruptions.”
In the other study, Shaul Hurwitz, a hydrogeologist at the U.S. Geological Survey in Menlo Park, and colleagues discovered CO2 and other dissolved gases such as nitrogen in many of Yellowstone’s other geysers. Understanding the role gas plays in steam eruptions is also important, Hurwitz says, because geysers aren’t the only way water erupts. The rapid boiling of an enclosed underground water reservoir can generate an explosive blast. In September 2014, a steam eruption rocked Japan’s Mount Ontake volcano without warning and killed 57 people. If CO2 helps fuel steam eruptions, then monitoring gas levels in groundwater could provide early warning, Hurwitz says.
As the weather warms, watch for falling rocks. While monitoring a cracked cliff in Yosemite National Park, researchers watched the fissure widen as temperatures rose. The risk of rockfalls could increase as climate change cranks the thermostat, one scientist predicts.
For three and a half years, geologists Brian Collins of the U.S. Geological Survey in Menlo Park, Calif., and Greg Stock of the National Park Service in Yosemite monitored a 19-meter-long crack in one of the park’s cliffs. The crack had a maximum width of about 12 centimeters. A measuring device anchored to both sides of the crack recorded changes in its size. The gap grew and shrank by as much as a centimeter daily as temperature changes caused the rock to expand and contract, the researchers report online March 28 in Nature Geoscience. Some effects lingered, however: The gap widened over the course of several summers and the constant size fluctuations further weakened the rock, the researchers say.
Around 25,000 tons of rocks and debris slipped down Yosemite’s slopes in 2015 — enough to fill more than three Olympic-sized swimming pools. About 15 percent of rockfalls from Yosemite’s granite cliffs occur during summer and at the hottest times of day. The rockfall risk could grow along with the cracks as the climate warms, geoscientist Valentin Gischig of ETH Zurich in Switzerland proposes in a perspective piece on the new finding.
Multiple sclerosis clue significant — A possible link between environment and multiple sclerosis (MS) could be a valuable tool in searching for the cause and cure of the disease…. Cases of MS seem to appear in clusters, and there is apparently some as yet unknown environmental factor that is distributed in the same way, reported Dr. John F. Kurtzke.… The highest frequency of MS is found in northern United States, southern Canada and northern Europe, where there are 30 to 60 cases per 100,000 population. — Science News, April 16, 1966
Update Researchers still aren’t sure what causes MS, a debilitating disease in which the body’s immune system attacks the insulation around nerve cell fibers. But research suggests that people who grow up farther from the equator, with reduced sun exposure, may have increased disease risk. The human body produces vitamin D in response to sunlight, and studies show that lower levels of vitamin D lead to higher MS risk (SN Online: 9/10/15). But other factors, including genetics and infections, may also play a role in disease development. Today, an estimated 90 MS cases occur for every 100,000 people in the United States.
The pale arch of light from the plane of our galaxy can be a humbling sight on a clear, dark night. But it’s just a sliver of all the treasures lurking in the Milky Way. Dense clouds of interstellar dust block visible light from remote regions of the galaxy but allow longer wavelengths to pass through. In February, astronomers completed a new map of our galaxy as seen in submillimeter light, which is shorter than radio waves but longer than infrared waves.
Submillimeter light can penetrate dust clouds, revealing details at the center of the galaxy and in stellar nurseries not visible at other wavelengths. The map was produced by ATLASGAL, a project using the APEX telescope in northern Chile to map part of the Milky Way. The project charted one-third of the band of galactic light that encircles our solar system; the images below show a narrow slice toward the constellation Sagittarius. Combined with images from the Spitzer and Planck satellites, the ATLASGAL map (top row) creates a detailed atlas of some of the cold structures in our galaxy. Dust clouds in places like the Trifid and Lagoon nebulas (circled, left), both a few thousand light-years away, glow faintly, as do filaments of detritus in the center of the galaxy (circled, right), 28,000 light-years from Earth. At near-infrared wave-lengths (center row), these regions nearly vanish behind obscuring curtains of dust. The galactic center remains hidden in visible light (bottom row) as well, though hot stars in Trifid and Lagoon radiate pools of hydrogen gas, making them glow.
A sugar that freshens air in rooms may also clean cholesterol out of hardened arteries.
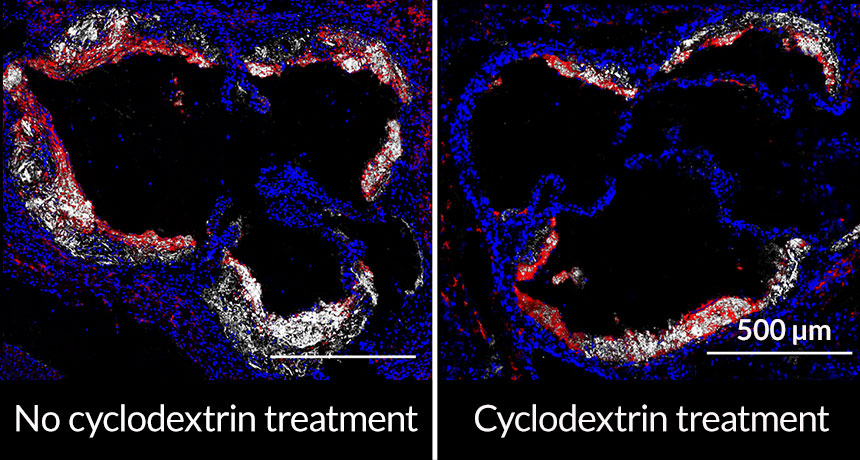
The sugar, cyclodextrin, removed cholesterol that had built up in the arteries of mice fed a high-fat diet, researchers report April 6 in Science Translational Medicine. The sugar enhances a natural cholesterol-removal process and persuades immune cells to soothe inflammation instead of provoking it, say immunologist Eicke Latz and colleagues.
Cyclodextrin, more formally known as 2-hydroxypropyl-beta-cyclodextrin, is the active ingredient in the air freshener Febreze. It is also used in a wide variety of drugs; it helps make hormones, antifungal chemicals, steroids and other compounds soluble. If the new results hold up in human studies, the sugar may also one day be used to liquefy cholesterol that clogs arteries. Other researchers say the approach is promising, but must be tested in clinical trials. The sweet molecule is generally considered safe, but injecting it may raise the risk of liver damage or hearing loss, says Elena Aikawa, a vascular biologist at Brigham and Women’s Hospital in Boston.
Mice taking cyclodextrin in the study did not exhibit side effects from the treatment, but previous work has indicated that the sugar may damage hearing in mice and cats. The molecule shunts cholesterol through the liver, so large cholesterol influxes might cause fat to build up in the liver, impairing its function. “Overall, cyclodextrin seems worth exploring as a therapeutic, although caution should be taken,” Aikawa says.
Cyclodextrin works by flipping a master switch, a gene called LXR, Latz and colleagues found. LXR’s protein turns on other genes involved in processing cholesterol and ushering it out of the body. The sugar also activated the LXR genes in human arteries examined in the lab and turned on inflammation-calming processes, Latz’s team discovered.
Latz, of the University Hospital Bonn in Germany, credits Nevada businesswoman Chris Hempel with the idea to use cyclodextrin to treat atherosclerosis. In people with the condition, cholesterol, calcium, immune cells and other substances form plaques inside arteries, hardening them. Plaques block blood flow and can break away and cause heart attacks and strokes (SN: 2/20/16, p. 32).
Hempel has twin daughters with a rare genetic disease known as Niemann-Pick Type C, in which cholesterol crystals clog organs, especially the brain. In 2009, the girls got special permission from the Food and Drug Administration for their doctor to give them infusions of cyclodextrin to dissolve the cholesterol crystals. Hempel later read a paper by Latz and colleagues in which the researchers described how cholesterol crystals irritate macrophages and provoke them to cause inflammation and heart disease. Macrophages normally patrol the body and help kill invading bacteria, viruses and other pathogens. The immune cells also gobble up cholesterol and deliver it to the liver where it can be made into bile and escorted out of the body in feces.
Hempel e-mailed Latz and suggested that cyclodextrin might melt the cholesterol crystals in arteries. Latz and his colleagues tested the idea by feeding mice genetically prone to atherosclerosis a high-fat diet and giving the animals regular injections of cyclodextrin under the skin. The sugar kept cholesterol plaques from building up in the rodents’ arteries. The scientists also found that cyclodextrin reduced already established plaques in mice by about 45 percent, even though the animals were still eating a high-fat diet.
Cyclodextrin could be used in combination with other drugs, such as statins, says Eran Elinav, an immunologist at the Weizmann Institute of Science in Rehovot, Israel. Statins and other drugs inhibit cholesterol production. “Potentially, combining cholesterol lowering with dissolution of preformed cholesterol in plaques could be additive,” Elinav says, “but this option needs to be explored in clinical trials.”
Although cyclodextrin is already approved by the FDA for use in people, it may be years before it’s known whether injecting the sugar will soften people’s hardened arteries. The sugar is not patentable, so no pharmaceutical companies have come forward to sponsor expensive clinical trials needed to get approval for this specific use, Latz says.